Organic electronics could change the electronics industry with their high cost-efficiencies and versatility compared with more commonly used inorganic electronics. For example, their flexibility could let companies print them like paper or put them into clothing to power wearable electronics. However, they have failed to gain much industry traction due to the difficulty of controlling their electronic structure.
To address this challenge, researchers at Argonne National Laboratory have developed a faster way of creating molecular models by using machine learning. The models dramatically cut the time needed to screen potential new organic materials for electronics.
The internal structure of an organic material affects its electrical efficiency. Current manufacturing processes for making these materials are sensitive, and the structures are extremely complex. This makes it difficult for scientists to predict the final structure and the material’s efficiency based on manufacturing conditions. For this, the Argonne team relies on machine learning—a way of training a computer to learn a pattern without being explicitly programmed to make those predictions.

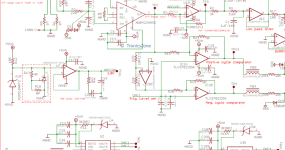
Schematic of the ANN-ECG method used in this work. Schematic example shows a three-bead/monomer coarse-grained molecular model mapping for sexi(3-methyl)thiophene. (Credit: Argonne National Laboratory)
The research focuses on using vapor deposition to assemble materials for organic electronics. In this process, scientists evaporate an organic molecule and let it slowly condense on a surface, producing a film. By manipulating certain deposition conditions, the scientists can finely tune the way the molecules form the film. The molecules can orient themselves in different ways, and the researchers want to know how structure influences the material’s electronic properties.
For example, they know that how molecules are arranged in the film affects the material’s charge mobility, a measure of how easily charges can move inside it. Charge mobility plays a role in the material’s efficiency as a device. To study this relationship and improve performance, the team runs detailed computer simulations of the vapor deposition process.
Some models simulate the behavior of all the electrons around each molecule at nanoscopic length and time scales. Naturally, these models are computationally intensive and take a long time to run.
To simulate the packing of entire devices, often containing millions of molecules, scientists must develop computationally cheaper, coarser models that describe the behavior of electrons in groups of molecules rather than individually. These coarse models can reduce computation time from hours to minutes, but the challenge is in making the coarse models truly predictive of the physical results. That‘s where machine learning comes into play: Its task is to uncover relationships between the detailed and coarse models.
Using an artificial neural network and a learning process called backpropagation, the machine learning algorithm learns to extrapolate from coarse to more detailed models. Using those complex relationships it finds between the models, it trains itself to make the same predictions on the electronic properties of the material using the coarse model as the detailed model would predict.
The resulting coarse model lets scientists screen two to three orders of magnitude more packing arrangements than before. Analysis results from the coarse model also help experimentalists more quickly develop high-performance materials.
Materials scientists have used machine learning before to find relationships between molecular structure and device performance, but this aims to do this by enhancing the interaction between models of different length and time scales.
Although the targeted goal of this research is to screen vapor deposited organic electronics, it could be used in many kinds of polymer research, and even fields such as protein science.
[“source=machinedesign”]